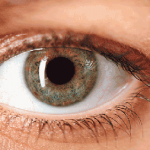
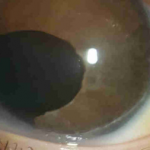
A síndrome de Axenfeld-Rieger (ARS) é um grupo clinicamente e geneticamente heterogêneo de desordens do desenvolvimento que afetam principalmente o segmento anterior do olho, conduzindo frequentemente a glaucoma secundário.
Pacientes com ARS também podem apresentar alterações sistêmicas, incluindo dismorfismo craniofacial leve e anomalias umbilicais. Caracteriza-se por herança autossômica dominante; o defeito subjacente em 40% dos pacientes é mutações em PITX ou FOXC. Sua incidência estimada é de 1: 200.000 em todo o mundo.
Manifestações oculares incluem embriotoxon posterior, alterações na íris e no ângulo anterior. Anomalias do desenvolvimento de anterior ângulo causa aumento da resistência ao fluxo e hipertensão ocular em quase 50% dos casos.
Os sinais craniofaciais característicos como hipoplasia maxilar, características dentárias incluindo hipodontia, oligodontia e microdontia e anomalias umbilicais também foram relatado em pacientes com RSA.
Características clínicas de pacientes com ARS
As manifestações clínicas da síndrome de Axenfeld-Rieger podem ser divididas em características oculares e sistêmicas.
Oculares
Pacientes apresentam malformações oculares, particularmente na íris, na córnea e no ângulo da câmara anterior.
O envolvimento ocular na síndrome é geralmente bilateral, no entanto, pode ser assimétrico ou, raramente, unilateral.
A corectopia é o deslocamento da pupila de sua posição normal, central, e é provável que seja associada com alta miopia ou ectopia lentis. Policoria é uma condição rara. As alterações da íris em pacientes com ARS podem variar de sutis a profundas.
Em alguns casos, com base na localização da pupila, corectopia e policoria contribuem para a intolerância anormal à percepção visual de luz (fotofobia). O deslocamento anterior da linha de Schwalbe causa embriotoxon posterior, uma córnea anormalidade geralmente herdada como um traço dominante e é um fator chave no diagnóstico de RSA.
O ocorrência de embriotoxon posterior em um paciente com um distúrbio do segmento anterior sem anormalidades da córnea, como opacidade da córnea, esclerocórnea ou megalocórnea, permitem distinguir ARS de outro transtorno do segmento anterior.
O glaucoma é a consequência mais grave da RSA e se desenvolve em aproximadamente 50% dos pacientes, que pode causar cegueira completa dentro de alguns anos. Bloqueio do canal de Schlemm, que é apenas um dos muitos fatores que resultam em glaucoma, causa o aumento do pressão, levando à morte das células ganglionares da retina e cegueira, se não tratada.
Sistêmicas
ARS também pode causar defeitos sistêmicos não oculares, incluindo anomalias dentárias, craniofacial dismorfismo e pele periumblical redundante.Os recursos faciais podem ser úteis ao sugerir diagnóstico de ARS, particularmente em membros da família com fenótipo ocular leve.
A base genética da ARS
Existem três tipos de ARS:
Os pacientes com ARS tipo 1 geralmente apresentam fenótipos oculares e sistêmicos. Anormalidades dentárias e faciais estão entre as características sistêmicas mais comuns dentro deste grupo.
Os pacientes com ARS tipo 2 comumente apresentam oligodontia, microdontia e perda prematura de dentes. Hipoplasia maxilar e defeitos umbilicais são menos comuns no tipo 2.
Os pacientes com ARS tipo 3 podem apresentar fenótipos oculares e sistêmicos, embora, mais tipicamente, apresentem apenas características oculares, especialmente disgenesia do segmento anterior, incluindo deslocamento anterior da linha de Schwalbe, hipoplasia do estroma da íris, corectopia e glaucoma. Na ARS tipo 3, pacientes raramente apresentam anomalias dentárias e dismorfismo facial e, ao invés disso, perda auditiva neurossensorial e anormalidades cardíacas parecem ser mais comuns.
Abordagem Diagnóstica
O diagnóstico de ARS é feito com base no exame oftalmológico e clínico, incluindo biomicroscopia do segmento anterior, medição da pressão intra-ocular, gonioscopia e oftalmoscopia. Baseado em clínica para os achados, um diagnóstico suspeito de ARS deve ser confirmado por testes genéticos, quando possível.
Tratamento
O tratamento atual de pacientes com ARS envolve principalmente o controle do glaucoma. Devido ao alto risco de glaucoma e por muitas vezes ser diagnosticado na infância, torna-se crucial seguir pacientes com ARS frequentemente para monitorização da PIO e do nervo óptico (relação escavação/disco).
Fonte: PEBMED